
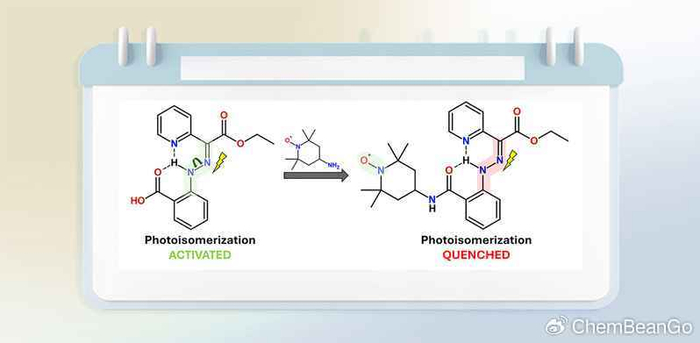
引言
腙官能团易于形成且稳定,在传感、抗菌、催化及作为分子开关等方面有广泛应用。其Z/E异构化能力使其对光和pH响应,并可用于构建超分子结构。腙在光分子应用中具有高热半衰期、高效光转换、抗疲劳等优点,被用于发光水凝胶、药物递送、分子执行器及太阳能储热器件。近期研究探索了腙开关与顺磁性物种结合,例如用于MRI造影剂。然而,利用电子顺磁共振研究自由基功能化腙开关的报道很少。近日,有研究报道了邻位卤素取代的吡啶转子腙具有光开关能力,引入了一种新型的邻位氮氧自由基取代的吡啶转子腙开关,旨在通过实验和理论方法研究其光开关性质。该研究发现其羧酸腙前体表现出部分可逆的光异构化,而自由基类似物的光异构化受到显著抑制。氮氧自由基的引入为利用EPR研究腙体系动力学提供了新机会。
进展
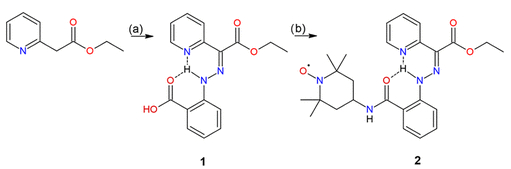
方案1. 制备化合物1和2的反应方案(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
化合物1通过Japp–Klingemann反应制备(方案1),经NMR验证。¹H NMR显示其以81% Z-型和19% E-型异构体混合物存在。化合物2通过CDI介导的4-氨基-TEMPO与1的酰胺化反应合成(方案1),产率59%。经MS和EPR表征,EPR谱显示典型的三线信号。
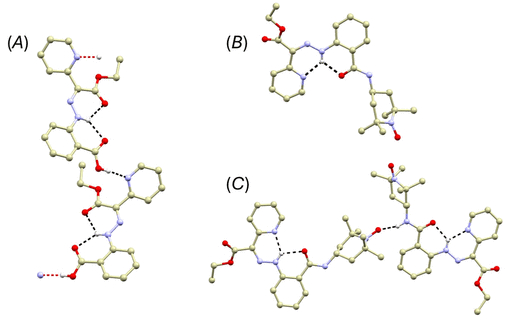
图1. (A) 化合物1晶体结构片段透视图。(B) 化合物2的分子结构。(C) 化合物2晶体结构片段透视图。(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
单晶X射线衍射显示,化合物1在固态下完全采取E-构象,通过两个分子内氢键稳定(图1A)。化合物2则完全采取Z-构象,腙N–H基团与吡啶氮及酰胺氧形成分叉分子内氢键(图1B, C)。使用量子理论中的原子理论分析表明,化合物2中的N–H···N氢键(10.2 kcal·mol⁻¹)强于化合物1中的相应氢键。
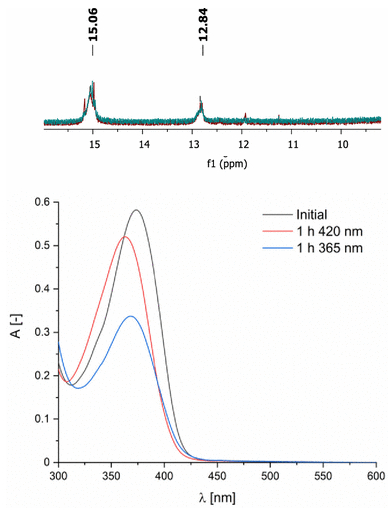
图2. 上图:化合物2在420 nm照射2小时前(绿色)后(红色)的¹H NMR谱中N–H共振峰的对比。下图:化合物2的UV–vis光谱在420 nm照射1小时和365 nm照射1小时前后的变化。(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
¹H NMR和UV–vis光谱表明,化合物1在420 nm和365 nm照射下发生可逆光异构化,达到光稳态时Z/E比分别为58/42和78/22。UV–vis光谱显示照射后吸收峰发生移动和强度变化。
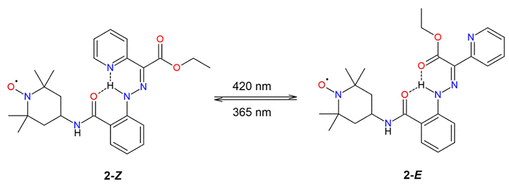
方案2. 考虑中的化合物2Z/E光异构化方案(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
相比之下,化合物2在420 nm长时间照射(2小时)后,¹H NMR谱未检测到Z/E比例变化(图2),表明其光异构化受到强烈抑制。UV–vis光谱显示,420 nm照射1小时引起吸收峰紫移和强度降低,而365 nm照射1小时导致显著的吸光度下降和光降解迹象(图2)。EPR光谱证实,420 nm照射后顺磁中心稳定,而365 nm照射后信号强度显著降低,可能与自由基分解有关。
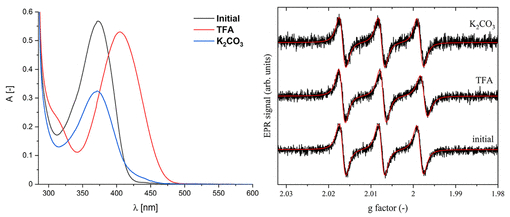
图3. 左图:化合物2在pH开关使用三氟乙酸和通过K₂CO₃塞过滤前后的UV–vis光谱。右图:化合物2在pH开关前后的X波段EPR谱(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
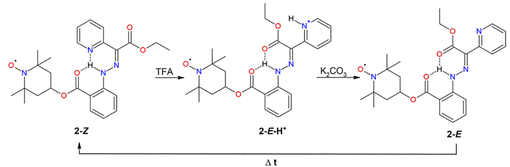
方案3. 化合物2在pH变化时(TFA=三氟乙酸)预期的Z/E异构化机制(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
化合物1和2均对pH变化响应。加入三氟乙酸(TFA)导致UV–vis光谱红移,颜色加深;经碳酸钾处理后恢复原状(图3,图S17)。对于化合物2,¹H NMR监测显示,TFA处理使E-异构体成为主要形式(67%),去质子化后恢复为Z-型为主(75%)(图S18)。EPR光谱显示,质子化后分子的旋转相关时间(τ_c)显著增加,表明分子迁移率降低;去质子化后τ_c部分恢复(图3,表S2)。
采用DFT和TD-DFT计算研究了化合物1和2的热异构化与光异构化机制。
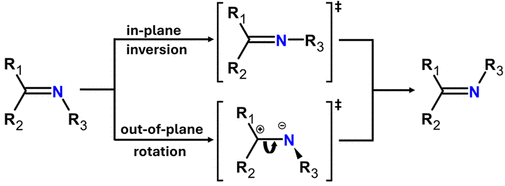
方案4. 腙的Z/E异构机制(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
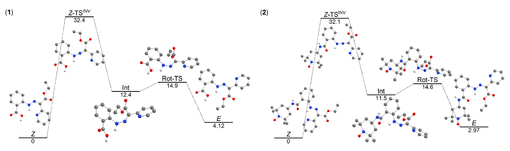
图4. DFT计算的Z→E异构化吉布斯能量剖面图(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
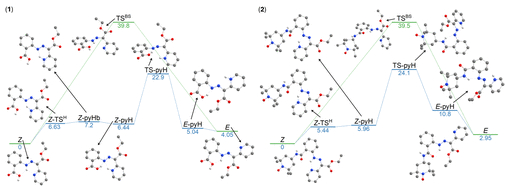
图5. DFT计算的Z→E异构化吉布斯能量剖面图(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
热异构化机制考察了平面内氮反转和平面外旋转(可能伴随质子转移)两种途径(方案4)。计算结果表明,两种化合物对于这两种热机制的活化吉布斯能垒(ΔG‡)相似,均在32 kcal·mol⁻¹左右或略低(图4,图5)。因此,热力学差异不能解释2的光异构化抑制。
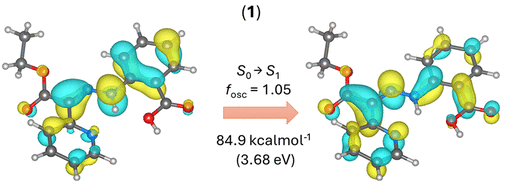
图6. 1-Z异构体的TD-DFT计算结果(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
TD-DFT计算显示,化合物1(Z型)的S₀→S₁跃迁(84.9 kcal·mol⁻¹)涉及腙部分的π→π*电子转移,有助于削弱C═N键,促进旋转异构化(图6)。
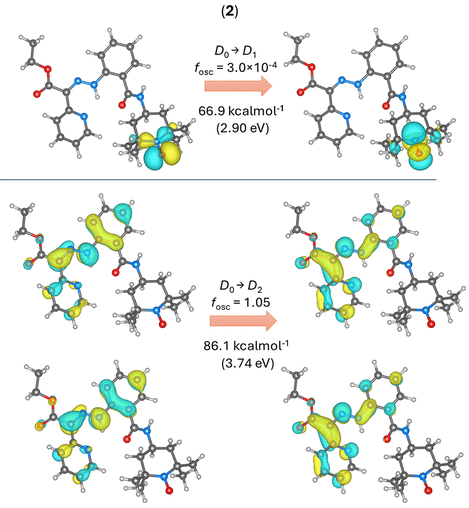
图7. 2-Z异构体的TD-DFT计算结果(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
对于化合物2(Z型),最低能量激发D₀→D₁跃迁(66.9 kcal·mol⁻¹)对应于TEMPO基团SOMO轨道内的π→π*转移,且振荡强度很弱(图7上)。下一个激发D₀→D₂(86.1 kcal·mol⁻¹)才涉及腙部分的π→π*转移,与化合物1的S₀→S₁类似(图7下)。
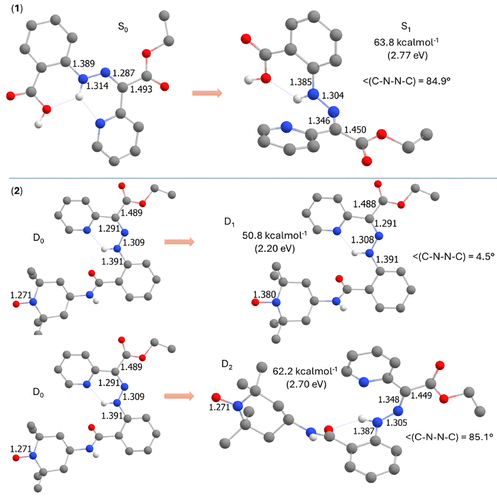
图8.1和2的Z异构体的TD-DFT计算结果,比较了基态和优化激发态的分子几何构型。(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
优化激发态几何结构发现,对于涉及腙激发的态(1的S₁和2的D₂),C═N键伸长、N–N键缩短,C–N–N–C二面角接近85°,表明该激发态利于异构化(图8)。
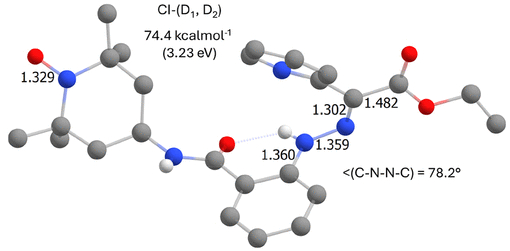
图9. 2-Z异构体的TD-DFT计算结果,显示两个激发态D₁和D₂之间锥形交叉点的分子几何构型。(图源:ACS Org. Inorg. Au2025,DOI: 10.1021/acsorginorgau.5c00068)
关键发现是,在化合物2中,搜索到了D₂和D₁激发态之间的锥形交叉点(图9)。从D₂到达此交叉点的能垒(约12.2 kcal·mol⁻¹)可能相对较低。因此,激发到D₂态的分子可能通过内转换迅速弛豫到D₁态,而D₁态(主要位于TEMPO基团)的几何变化不影响腙键,从而导致光异构化过程被淬灭。这为化合物2光异构化受抑制提供了合理解释。
结论
该研究成功合成并表征了一种新型氮氧自由基取代的腙开关。其实验研究表明,与羧酸前体1不同,化合物2的光诱导Z/E异构化受到显著抑制,这主要归因于TEMPO自由基的引入。理论计算揭示,化合物2中存在的与自由基相关的低能激发态(D₁)可能导致从腙局部激发态(D₂)发生快速内转换,从而猝灭了光异构化途径。另一方面,化合物2仍表现出可逆的pH响应异构化。该研究为理解自由基修饰对光开关分子性能的影响提供了新见解,并指出未来设计此类体系时,需确保最低激发态位于腙开关部分而非自由基基团上。氮氧自由基的引入为利用EPR光谱研究腙体系的动力学(如pH响应过程)提供了新的可能性。
【声明】本文在创作过程中,笔者提供了创作思路,部分内容使用AI工具辅助优化,并经过笔者修改润色。本文内容仅代表个人观点,旨在进行知识分享和行业探讨。

